
Какие соединения проявляют как кислотные так и основные свойства

Периодический закон был открыт Д.И. Менделеевым в 1868 году. Его современная формулировка: свойства химических элементов и образуемых ими
соединений (простых и сложных) находятся в периодической зависимости от величины заряда атомного ядра.
Периодический закон лежит в основе современного учения о строении вещества. Периодическая система Д.И. Менделеева является наглядным отражением
периодического закона.
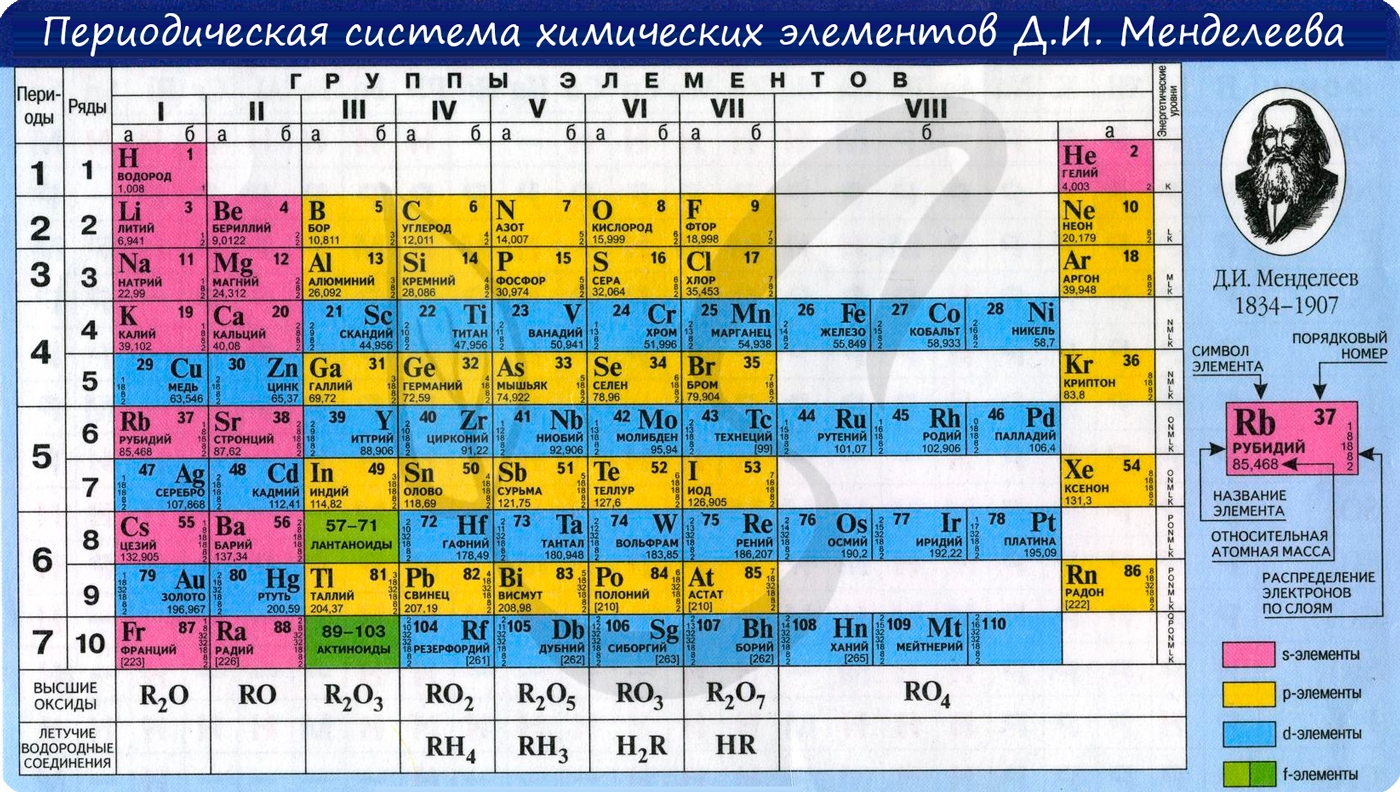
В периодической таблице элементы расположены в порядке увеличения атомного заряда, группируются в “строки и столбцы” – периоды и группы.
Период – ряд горизонтально расположенных химических элементов. 1, 2 и 3 периоды называются малыми, они состоят из одного ряда элементов.
4, 5, 6 – называются большими периодами, они состоят из двух рядов химических элементов.
Группой называют вертикальный ряд химических элементов в периодической таблице. Элементы собраны в группы на основе степени окисления в
высшем оксиде. Каждая из восьми групп состоит из главной подгруппы (а) и побочной подгруппы (б).
Периодическая таблица Д.И. Менделеева содержит колоссальное число ответов на самые разные вопросы. При умелом ее использовании вы сможете
предполагать строение и свойства веществ, успешно писать химические реакции и решать задачи.

Радиус атома
Радиусом атома называют расстояние между атомным ядром и самой дальней электронной орбиталью. Это не четкая, а условная граница, которая
говорит о наиболее вероятном месте нахождения электрона.
В периоде радиус атома уменьшается с увеличением порядкового номера элементов (“→” слева направо). Это связано с тем, что с увеличением номера группы
увеличивается число электронов на внешнем уровне. Запомните, что для элементов главных подгрупп номер группы равен числу электронов на внешнем уровне.
С увеличением числа электронов они становятся более скученными, так как притягиваются друг к другу сильнее: это и есть причина маленького радиуса атома.
Чем меньше электронов, тем больше у них свободы и больше радиус атома, поэтому радиус увеличивается в периоде “←” справа налево.

В группе радиус атома увеличивается с увеличением заряда атомных ядер – сверху вниз “↓”. Чем больше период, тем больше электронных орбиталей вокруг атома,
соответственно, и больше его радиус.
С уменьшением заряда атома в группе радиус атома уменьшается – снизу вверх “↑”. Это связано с уменьшением количества электронных орбиталей вокруг
атома. Для примера возьмем атомы бора и алюминия, элементов, расположенных в одной группе.
Период, группа и электронная конфигурация
Обратите внимание еще раз на важную деталь: элементы, находящиеся в одной группе (главной подгруппе!), имеют сходную конфигурацию внешнего уровня.
Так у бора на внешнем уровне расположены 3 электрона, у алюминия – тоже 3. Оба они в III группе.
Такая закономерность иногда может сильно облегчить жизнь, однако у элементов побочных подгрупп она отсутствует – там нужно считать электроны
“вручную”, располагая их на электронных орбиталях.
Раз уж мы повели речь об электронных конфигурациях, давайте запишем их для бора и алюминия, чтобы лучше представлять их внешний уровень и увидеть
то самое “сходство”:
- B5 – 1s22s22p1
- Al13 – 1s22s22p63s23p1
Общую электронную конфигурацию для элементов III группы главной подгруппы можно записать ns2np1. Это будет работать для
бора, внешний уровень которого 2s22p1, алюминия – 3s23p1, галия – 4s24p1,
индия – 5s25p1 и таллия – 6s26p1. За “n” мы принимаем номер периода.
Правило составления электронной конфигурации, которое вы только что увидели, универсально. Если вы имеете дело с элементом главной подгруппы,
то увидев номер группы вы знаете, сколько электронов у него на внешнем уровне. Посмотрев на период, знаете номер его внешнего уровня.
Вам остается только распределить известное число электронов по s и p ячейкам, а затем подставить номер периода – и вот быстро получена
конфигурация внешнего уровня. Предлагаю посмотреть на примере ниже 🙂

Очень надеюсь, что теперь вы знаете: только глядя на положение элемента в периодической таблице, на группу и период, в которых он расположен,
вы уже можете составить конфигурацию его внешнего уровня. Безусловно, это для элементов главных подгрупп. Повторюсь: у побочных – только “вручную”.
Длина связи
Длина связи – расстояние между атомами химически связанных элементов. Очевидно, что понятия длины связи и атомного радиуса взаимосвязаны напрямую.
Чем больше радиус атома, тем больше длина связи.
Убедимся в этом на наглядном примере, сравнив длину связей в четырех веществах: HF, HCl, HBr, HI.
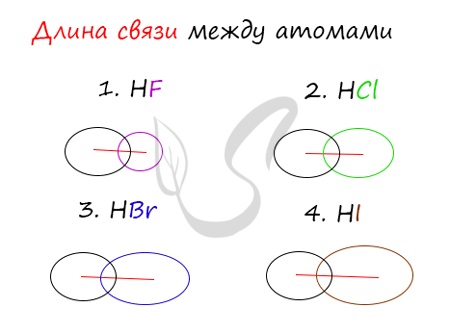
Чем больше радиусы атомов, которые образуют химическую связь, тем больше между ними и длина связи. Радиус атома водорода неизменен во всех трех
веществах, а в ряду F → Cl → Br → I происходит увеличение радиуса атома. Наибольшим радиусом обладает йод, поэтому самая длинная связь в молекуле HI.
Металлические и неметаллические свойства
В периоде с увеличением заряда атома металлические свойства ослабевают, неметаллические – усиливаются (слева направо “→”). В группе с увеличением
заряда атома металлические свойства усиливаются, а неметаллические – ослабевают (сверху вниз “↓”).

Сравним металлические и неметаллические свойства Rb, Na, Al, S. Натрий, алюминий и сера находятся в одном периоде. Металлические свойства возрастают
S → Al → Na. Натрий и рубидий находятся в одной группе, металлические свойства возрастают Na → Rb.
Таким образом, самые сильные металлические свойства проявляет рубидий, но с другой стороны – у него самые слабые неметаллические свойства. Сера
обладает самыми слабыми металлическими свойствами, но, если посмотреть по-другому, сера – самый сильный неметалл.
Распределение металлов и неметаллов в периодической таблице также является наглядным отображением этого правила. Если провести условную
линию, проходящую от бора до астата, то справа окажутся неметаллы, а слева – металлы.

Основные и кислотные свойства
Основные свойства в периоде с увеличением заряда атома уменьшаются, кислотные – возрастают. В группе с увеличением заряда атома основные
свойства усиливаются, а кислотные – ослабевают.
Кислотные и основные свойства противопоставлены друг другу, как противопоставлены металлические и неметаллические. Где первые усиливаются,
вторые – убывают. Все аналогично, поэтому смело ассоциируйте одни с другими, так будет гораздо легче запомнить.

Замечу, что здесь есть одно важное исключение. Как и в общем случае: исключения только подтверждают правила. В ряду галогенводородных
кислот HF → HCl → HBr → HI происходит усиление кислотных свойств (а не ослабление, как должно быть по логике нашего правила).
Это можно объяснить в темах диссоциации и химических связей. Когда мы дойдем до соответствующей темы, я напомню про HF и водородные связи между
молекулами, которые делают эту кислоту самой слабой. Сейчас воспринимайте это как исключение: HF – самая слабая из этих кислот, а
HI – самая сильная.

Восстановительные и окислительные свойства
Восстановительные свойства в периоде с увеличением заряда атома ослабевают, окислительные – усиливаются. В группе с увеличением заряда
атома восстановительные свойства усиливаются, а окислительные – ослабевают.
Ассоциируйте восстановительные свойства с металлическими и основными, а окислительные – с неметаллическими и кислотными. Так гораздо проще
запомнить 😉

Электроотрицательность (ЭО), энергия связи, ионизации и сродства к электрону
Электроотрицательность – способность атома, связанного с другими, приобретать отрицательный заряд (притягивать к себе электроны).
Мы уже касались ее в статье, посвященной степени окисления. Это важное свойство, ведь более ЭО-ый атом притягивает
к себе электроны и уходит в отрицательную степень окисления со знаком минус “-“.
Все перечисленные в подзаголовке свойства вместе с ЭО усиливаются в периоде с увеличением заряда атома, в группе с увеличением заряда атома
они ослабевают. Таким образом, самый электроотрицательный элемент расположен справа вверху таблицы Д.И. Менделеева – это фтор.

Для примера сравним ЭО-ость атомов Te, In, Al, P. Индий расположен в одной группе с алюминием, ЭО-ость In → Al возрастает (снизу вверх). Алюминий
расположен в одном периоде с серой, ЭО-ость возрастает Al → S (слева направо). Сравнивая серу и теллур, мы видим, что сера расположена в группе
выше теллура, значит и ее электроотрицательность тоже выше.
Энергия связи (а также ее прочность) возрастают с увеличением электроотрицательности атомов, образующих данную связь. Чем сильнее атом тянет на
себя электроны (чем больше он ЭО-ый), тем прочнее получается связь, которую он образует.
Понятию ЭО-ости “синонимичны” также понятия сродства к электрону – энергии, выделяющейся при присоединении электрона к атому, и энергии ионизации –
количеству энергии, которое необходимо для отщепления электрона от атома. И то, и другое возрастают с увеличением электроотрицательности.
Продемонстрирую на примере. Сравним энергию связи в трех молекулах: H2O, H2S, H2Se.

Высшие оксиды и летучие водородные соединения (ЛВС)
В периодической таблице Д.И. Менделеева ниже 7 периода находится строка, в которой для каждой группы указаны соответствующие высшие оксиды,
ниже строка с летучими водородными соединениями.
Для элементов главных подгрупп начиная с IV группы (в большинстве случае) максимальная степень окисления (СО) определяется по номеру группы. К примеру,
для серы (в VI группе) максимальная СО = +6, которую она проявляет в соединениях: H2SO4, SO3.
В таблице видно, что для VIa группы формула высшего оксида RO3, а, к примеру, для IIIa группы – R2O3. Напишем
высшие оксиды для веществ из VIa : SO3, SeO3, TeO3 и IIIa группы: B2O3, Al2O3,
Ga2O3.
На экзамене строка с готовыми “высшими” оксидами, как в таблице наверху, может отсутствовать. Считаю важным подготовить вас к этому. Предположим,
что эта строчка внезапно исчезла из таблицы, и вам нужно записать высшие оксиды для фосфора и углерода.

С летучими водородными соединениями (ЛВС) ситуация аналогичная: их может не быть в периодической таблице Д.И. Менделеева, которая попадется на экзамене.
Я расскажу вам, как легко их запомнить.
ЛВС характерны для IV, V, VI и VII группы. Элементы этих групп более электроотрицательны, чем водород, поэтому ходят в “-” отрицательную СО.
Минимальная степень окисления для элементов главных подгрупп, начиная с IV группы, может быть рассчитана так: номер группы – 8.
Например, для углерода минимальная СО = 4-8 = -4; для азота 5-8 = -3; для кислорода 6-8 = -2; для фтора 7-8 = -1. Для того, чтобы запомнить
ЛВС, вы должны ассоциировать IV, V, VI и VII группы с хорошо известными вам веществами: метаном, аммиаком, водой и фтороводородом.

Так как общее строение ЛВС в пределах одной группы сходно, то, вспомнив например H2O для кислорода в VI группе, вы легко
найдете формулы других ЛВС VI группы: серы – H2S, H2Se, H2Te, H2Po.
© Беллевич Юрий Сергеевич 2018-2020
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение
(в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов
без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования,
обратитесь, пожалуйста, к Беллевичу Юрию.
Источник
Кислотные и основные свойства органических соединений являются важными аспектами их реакционной способности. Перенос протона водорода наблюдается в ходе многих биохимических реакций.
Современные представления о природе кислот и оснований основаны на теории Бренстеда.
По Бренстеду, кислоты – это нейтральные молекулы и ионы, способные отдавать протоны водорода (доноры протонов). Основаниями являются нейтральные молекулы и ионы, способные присоединять протоны водорода (акцепторы протонов).
Кислотно-основное взаимодействие может быть представлено схемой:
A-H + B A- + B+-H
кислота основание сопряжённые
основание кислота
Кислота A-H, теряя протон, превращается в основание A-, которое называют сопряжённым основанием данной кислоты. Наоборот, основание B, принимая протон, превращается в сопряжённую кислоту B+-H.
В кислотно-основном равновесии существует важная закономерность: чем сильнее кислота, тем слабее сопряжённое с ней основание.
Понятие кислотности и основности взаимосвязаны: кислотные свойства проявляются только в присутствии оснований и наоборот.
Например, газообразный хлороводород не проявляет кислотных свойств, для этого необходимо присутствие основания:
HCl + H2O Cl – + H3O+
кислота основание сопряжённые
основание кислота
Некоторые соединения в зависимости от условий могут проявлять и кислотные, и основные свойства, т.е. понятия «кислота» и «основание» относительны. Например, в присутствии воды как основания уксусная кислота проявляет кислотные свойства, а в присутствии концентрированной серной кислоты – основные:
Так как в биохимических процессах растворителем обычно является вода, то дальше мы будем рассуждать о кислотно-основных свойствах соединений по отношению к воде.
Кислоты Бренстеда
Сила кислот количественно выражается константой равновесия реакции (К), заключающейся в переносе протона водорода от кислоты к основанию – молекуле воды.
Используя значение константы равновесия этой реакции и учитывая, что концентрация воды практически постоянна, можно определить произведение K.[H2O], называемое константой кислотности (Ka) (от слова acid – кислота).
Чем больше Ka, тем сильнее кислота. Но величины Ka очень малы (например, Ka уксусной кислоты 1,75. 10-5 ), пользоваться ими неудобно, поэтому введено понятие «показатель константы кислотности» (её отрицательный логарифм) – pKa. pKa=-lg Ka. Например, для уксусной кислоты pKa=4,75. Чем меньше величина pKa, тем сильнее кислота.
В зависимости природы кислотного центра (атома, отщепляющего протон водорода) различают следующие типы кислот:
– OH-кислоты (вода, спирты, фенолы, карбоновые кислоты);
– SH-кислоты (тиоспирты, тиофенолы, тиокислоты);
– NH-кислоты (аммиак, амины, амиды кислот, пиррол);
– CH-кислоты (углеводороды и их производные).
Сила кислот определяется стабильностью сопряжённых оснований – чем стабильнее сопряжённое основание (анион кислоты), тем сильнее кислота.
Стабильность сопряжённого основания определяется степенью делокализации заряда, которая зависит от следующих факторов: природа атома в кислотном центре, влияние радикала, степень сольватации.
Рассмотрим каждый из этих факторов.
Природа атома в кислотном центре. Для делокализации отрицательного заряда наибольшее значение имеют электроотрицательность атома и его поляризуемость.
Чем выше электроотрицательность атома, получившего отрицательный заряд после отщепления протона, тем сильнее он этот отрицательный заряд удерживает и труднее предоставляет протону, а значит равновесие A-H А- + H+смещается вправо.
В периодах слева направо электроотрицательность возрастает. Поэтому OH-кислоты сильнее NH-кислот, а NH-кислоты сильнее CH-кислот (при прочих равных условиях).
Так как электроотрицательность атомов зависит от типа гибридизации, то среди CH-кислот при переходе от алканов к алкенам и далее к алкинам кислотность возрастает:
CH3-CH3 < CH2=CH2 < CH≡CH.
Так, этан и этилен не реагируют с амидом натрия, а ацетилен – реагирует:
В группах сверху вниз увеличивается поляризуемость. Это связано с тем, что увеличивается число энергетических уровней, т.е. объем электронных оболочек. Чем выше поляризуемость, тем более делокализован заряд, тем стабильнее анион, а значит, выше сила кислоты. Поэтому SH-кислоты сильнее, чем OH-кислоты. Например, спирты не реагируют со щелочами, а тиолы – реагируют:
Влияние радикала (заместителя). Кислотность органических соединений в значительной степени зависит от природы заместителя, связанного с реакционным центром. Особенно сильно повышает кислотные свойства наличие сопряженной системы в анионе.
Сравним кислотные свойства спиртов и фенолов.
В случае фенолят-аниона (p,π-сопряженная система) степень делокализации заряда гораздо выше, поэтому фенолы более сильные OH-кислоты, чем спирты. Например, фенолы в отличие от спиртов реагируют с щелочами:
Разницу в кислотных свойствах можно подтвердить и величинами pKa: для фенола pKa=10,00; для метанола pKa=16,00.
Фенолы – более сильные кислоты, чем спирты, но и они являются слабыми: фенол не вытесняет угольную кислоту из её солей, т.е. является более слабой кислотой, чем угольная.
Сравнивая кислотные свойства фенолов и карбоновых кислот, нужно отметить, что и фенолят-, и ацилат-анион являются p,π-сопряженными системами, но в ацилат-анионе наблюдается более полная делокализация отрицательного заряда за счёт двух электроотрицательных атомов кислорода:
В отличие от фенолов карбоновые кислоты взаимодействуют с гидрокарбонатом натрия:
, т.е. они явля-
ются более сильными кислотами, чем угольная кислота.
Заместители в радикале также влияют на кислотные свойства. Если заместитель способствует делокализации отрицательного заряда в анионе (электроноакцепторный заместитель), то кислотность будет выше. И наоборот, электронодонорные заместители затрудняют делокализацию заряда и поэтому понижают кислотные свойства.
Сравним кислотные свойства следующих соединений: масляная кислота, α-хлормасляная и β-хлормасляные кислоты.
Анион масляной кислоты стабилизируется только за счёт слабого индуктивного эффекта углеводородного радикала. Атом хлора проявляет сильный отрицательный индуктивный эффект, поэтому гораздо более сильно стабилизирует анион кислоты. Так как индуктивный эффект передается с затуханием, влияние хлора в α-положении проявляется сильнее. Итак, самой сильной является α-хлормасляная кислота, затем – β-хлормасляная и, наконец, самая слабая кислота – масляная. Это подтверждается и значениями pKa данных кислот.
В ароматическом ряду электроноакцепторные заместители также способствуют делокализации заряда, таким образом увеличивая кислотные свойства. Электронодонорные заместители оказывают обратное влияние.
pKa=7,16 pKa=10,0 pKa=10,68
Суммируя все наши рассуждения, можно выстроить ряд убывания кислотных свойств OH-кислот:
карбоновые кислоты > угольная кислота > фенолы > спирты.
(Причем в каждой группе электронодонорные заместители понижают кислотность, а электроноакцепторные – повышают).
Основания Бренстеда
Основания, по Бренстеду, это нейтральные молекулы и ионы, способные присоединять протон водорода. Для образования ковалентной связи с протоном основания Бренстеда должны предоставлять или неподелённую электронную пару, или электроны π-связи. В соответствии с этим основания Бренстеда делятся на n-основания и π-основания.
n-Основания – это анионы или нейтральные молекулы, имеющие атом с неподелённой электронной парой. Их классифицируют по центрам основности следующим образом:
– оксониевые основания: спирты R-ÖH, простые эфиры
R-Ö-R, сложные эфиры , альдегиды и кетоны
– аммониевые основания: амины , гетероциклические соединения, например, пиридин
– сульфониевые основания: тиоспирты , тиоэфиры .
π-Основания – соединения, имеющие π-связи, т.е. алкены, алкадиены, алкины, арены. Это очень слабые основания, т.к. протонируемые электронные пары несвободны. Например, этен проявляет π-основные свойства при образовании π-комплекса с протоном водорода:
Для количественной характеристики основности используют величину pKa сопряжённой с данным основанием кислоты (BH+). Эту величину обозначают pKBH+. Чем pKBH+ больше, тем сильнее основание.
Влияние природы атома в основном центре и связанных с ним заместителей на основность противоположно рассмотренному ранее их влиянию на кислотность:
– с увеличением электроотрицательности атома основного центра основность уменьшается (атом труднее отдаёт свою неподелённую электронную пару для присоединения протона), т.е. аммониевые основания сильнее оксониевых. Так, этанол способен взаимодействовать только с концентрированными минеральными кислотами:
,
а этиламин проявляет основные свойства даже при взаимодействии с водой:
.
– с увеличением поляризуемости атома основного центра основность уменьшается, т.е. оксониевые основания сильнее сульфониевых;
– электронодонорные заместители повышают основность, а электроноакцепторные – понижают (чем выше электронная плотность на основном центре, тем легче он предоставит свою электронную пару протону). Так, в ряду п-нитроанилин, анилин,
п-толуидин основность повышается: нитро-группа является электроноакцепторным заместителем, а метильная группа – электронодонорным. Это подтверждают и значения pKBH+.
pKBH+=1,00 pKBH+=4,60 pKBH+=5,10
Итак, самыми сильными основаниями являются аммониевые. Основность различных типов аминов мы обсудим, рассматривая химические свойства аминов в целом.
АМИНЫ
Амины – это производные аммиака, в молекуле которого один, два или три атома водорода замещены углеводородными радикалами. Отсюда первый тип классификации аминов: по количеству радикалов амины подразделяют на первичные, вторичные и третичные.
Другой вид классификации аминов – по природе радикалов. Амины подразделяют на алифатические и ароматические. Например, приведенные выше амины являются алифатическими, а анилин (аминобензол) – ароматическим:
Для названия аминов применяют радикало-функциональную и заместительную номенклатуру IUPAC. По радикало-функциональной номенклатуре называют радикал или радикалы, если их несколько (в алфавитном порядке), и добавляют слово «амин», например, метиламин, метилэтиламин, диметиламин и т.д. Те амины, которые нельзя назвать по радикало-функциональной номенклатуре (сложные радикалы), называют по заместительной номенклатуре, например:
Ароматические амины обычно рассматривают как производные анилина, например:
За счёт неподелённой электронной пары азота амины проявляют основные и нуклеофильные свойства.
Сравним основные свойства различных групп аминов.
Алифатические амины являются более сильными основаниями, чем ароматические. Это связано с тем, что неподелённая электронная пара азота в ароматических аминах участвует в p,π-сопряжении и менее доступна для атаки протона водорода. В алифатических же аминах электронная плотность на атоме азота аминогруппы повышена за счёт электронодонорного влияния алкильных групп:
Алифатические амины взаимодействуют с минеральными кислотами, карбоновыми кислотами и даже с водой (очень слабой кислотой):
Ароматические амины как слабые основания взаимодействуют с минеральными кислотами:
Заместители в ароматическом кольце влияют на основные свойства аминов: электронодонорные заместители повышают основные свойства, а электроноакцепторные – понижают (см. стр.71).
Сравним основные свойства различных типов алифатических аминов – первичных, вторичных и третичных, например, метиламина, диметиламина и триметиламина.
Чем выше электронная плотность на атоме азота, тем выше основные свойства амина. Каждая метильная группа смещает электронную плотность к атому азота, поэтому можно было бы предположить, что самым сильным основанием является третичный амин. Однако, сравнивая значения pKBH+, можно увидеть, что это не так: самым сильным основанием является вторичный амин. Этот факт можно объяснить с позиций пространственной доступности неподелённой электронной пары азота: в триметиламине три крупных заместителя «прикрывают» её. Таким образом, ряд убывания основности алифатических аминов:
вторичные > первичные > третичные.
За счет неподелённой электронной пары азота амины проявляют также нуклеофильные свойства. Амины являются нуклеофилами, например, в реакциях алкилирования и ацилирования.
Алкилирование – это введение в молекулу алкила (метил, этил, пропил и т.п.). В качестве алкилирующих реагентов обычно используют алкилгалогениды (этилхлорид, метилбромид, например). Так, в реакции этиламина с метилхлоридом образуется метилэтиламин:
Это реакция нуклеофильного замещения. Она позволяет получить вторичный амин из первичного и третичный – из вторичного.
Ароматические амины также вступают в реакции алкилирования, но менее активно, т.к. их нуклеофильные свойства понижены (неподелённая электронная пара, отвечающая за них, участвует в p,π-сопряжении).
Ацилированием называют введение в молекулу ацила – остатка карбоновой кислоты (например, ацетил – это остаток уксусной кислоты, пропионил – пропионовой). Для ацилирования аминов обычно используют ангидриды соответствующих кислот.
Подробнее мы будем рассматривать эти реакции, изучая тему «Карбоновые кислоты и их функциональные производные».
Для ароматических аминов характерны также реакции, протекающие за счёт ароматического кольца – реакции электрофильного замещения. Ранее мы уже рассматривали влияние аминогруппы на ход реакций SE (см. стр.44,45): аминогруппа, являясь электронодонорным заместителем (+MNH2 >> -INH2), облегчает эти реакции в сравнении с бензолом и является орто-, пара-ориентантом. Например, бромирование анилина бромной водой приводит к образованию белого осадка 2,4,6-триброманилина:
Эту реакцию используют для качественного обнаружения анилина.
Источник